By Mark Plavsic
 Use of continuous cell lines in the manufacture of biological therapeutic products, such as vaccines, recombinant proteins, and monoclonal antibodies, is associated with the concomitant risk of process/product contamination with endogenous retroviruses, latent viruses, or new and emerging adventitious agents. Cell-culture applications are impossible without the use of nutrient media for cell multiplication and subsequent product generation. Although several serum-free and chemically defined nutrient media formulations are available for commercial use, many cell-culture applications require use of nutrient media supplementation with serum or other animal-derived components. Use of serum-supplemented cell-culture media is considered a point of entry for the introduction of adventitious agents into a manufacturing process. Other raw materials used in the manufacture of biotherapeutics, especially those of animal and human origin, could also present a viral safety risk.
Use of continuous cell lines in the manufacture of biological therapeutic products, such as vaccines, recombinant proteins, and monoclonal antibodies, is associated with the concomitant risk of process/product contamination with endogenous retroviruses, latent viruses, or new and emerging adventitious agents. Cell-culture applications are impossible without the use of nutrient media for cell multiplication and subsequent product generation. Although several serum-free and chemically defined nutrient media formulations are available for commercial use, many cell-culture applications require use of nutrient media supplementation with serum or other animal-derived components. Use of serum-supplemented cell-culture media is considered a point of entry for the introduction of adventitious agents into a manufacturing process. Other raw materials used in the manufacture of biotherapeutics, especially those of animal and human origin, could also present a viral safety risk.
Traditionally, the management of inadvertent virus contamination is achieved through the incorporation of various measures aimed to preclude, detect, and inactivate adventitious viral agents from the biological products (i.e., selection, testing, and clearance).
Although a plethora of regulatory guidance documents have been enacted governing product safety from adventitious agents (1–16), complete risk elimination has not yet been achieved. Several examples of bioproduction process contamination have been documented over the years, implicating minute virus of mice (MVM), epizootic hemorrhagic disease virus (EHDV), reovirus 2 (REO-2), cache valley virus (CVV), and calicivirus 2117 (17–23). Instances of final product contamination with adventitious agents have also been published (24, 25).
A recent Pharma IQ study (26) revealed that 37.5% of respondents—who were from the biotechnology manufacturing industry in the United States and Europe—named viruses as their biggest concern, despite the fact that nearly two thirds (62.5%) of surveyed participants already had strategies in place to mitigate the risk of viral contamination. More than a third (37.5%) of participants said their organization was satisfied with the solutions they currently have in place. These strategies so far appear to be working on the whole, as 87.5% of companies also reported they have not had to deal with a contamination.
The purpose of this article is to discuss some holistic, interlocking approaches across the manufacturing chain to reduce the risk of adventitious viral agent contamination and to ensure uninterrupted supply of safe biological products to patients in need.
Product safety and quality by design (QbD)
Traditionally, product safety has relied on the incorporation of three key measures into the manufacturing process: selection, testing, and viral clearance. These measures are collectively known as the “safety triangle” (Figure 1).
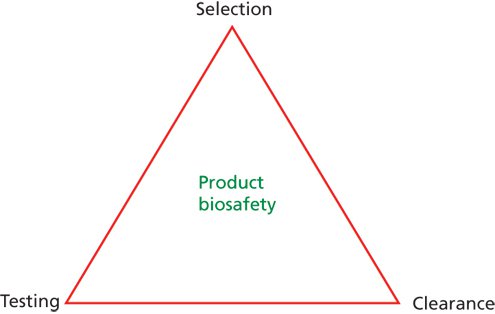 Figure 1: Illustration of the biotherapeutics safety triangle.
Figure 1: Illustration of the biotherapeutics safety triangle.
The elements of the safety triangle include the selection of source materials and release based on prior material and supplier quality approval and qualification testing for adventitious contaminants; testing for various adventitious contaminants at appropriate stages of the manufacturing process from raw materials, starting materials (e.g., cell banks, viral and bacterial seeds), and manufacturing intermediates; and viral clearance, employed either in raw material control or evaluation of the capacity and capability of the downstream purification process to clear (remove or inactivate) potential adventitious contaminants.
Although the safety triangle still represents a central dogma in the viral safety of biological products, it has been generally accepted that the safety triangle alone may not be sufficient, and some enhancements may be warranted.
Today’s industry and regulatory expectations require that an effective viral risk mitigation strategy be built into the whole manufacturing chain, spanning from the suppliers of crucial raw materials to the fill/finish and contract manufacturing organizations (CMO), where applicable (Figure 2).
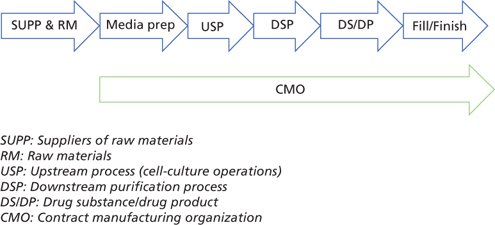
Figure 2: Effective viral risk mitigation across the whole manufacturing process (example: r-proteins, a typical recombinant protein manufacturing process)
In this context, viral risk mitigation should be an integral part of the overall quality system and quality risk management strategy (12). Viral safety needs to be designed into the overall drug-development process and QbD approach.
The concept of product “safety by design” (SbD) represents an integrated, holistic approach to viral safety across the manufacturing chain. The goal of SbD is to protect a manufacturing process from interruptions caused by viral contamination and ensure product and patient safety by preventing virus introduction, ensuring early detection, and enabling rapid response to ensure containment/elimination of viruses if introduced into the manufacturing process. It typically spans the following five areas:
• Raw materials (RM)
• New process/product development (PD)
• Manufacturing process
• Quality system (QS)
• Detection (testing).
In the context of this article, animal-origin (AO) and chemically defined (CD) raw materials are defined as follows.
Animal (including human) origin materials are derived from various species of animals, including humans, and they can be either primary (direct) or secondary (indirect).
Primary (direct) animal origin materials are derived from animals or their tissues with or without further processing. Unprocessed primary animal origin materials are derived with minimal or no further processing. Examples include: whole blood, serum, cells, tissue extracts, and intestinal mucosa. Processed primary animal origin materials are derived after a series of processing steps such as extraction, precipitation, or purification. Examples include: bovine serum albumin (BSA), human serum albumin (HSA), porcine trypsin, purified enzymes, wool-derived cholesterol, heparin, gelatin, casein, and collagen.
Secondary (indirect) animal origin materials are recombinant proteins derived from manufacturing (e.g., fermentation) processes where animal-origin materials were added.
Chemically defined raw materials have all components identified; components are in the chemical form, and the structure and concentration of all components is known and of high purity with only minimal levels of trace chemical impurities.
The following sections address viral risk mitigation across the five areas in more detail.
Raw materials
Raw materials have been regarded as one of the main portals of viral entry into a GMP manufacturing environment. The main goal of viral risk mitigation at this level is to prevent virus introduction into a manufacturing process via raw materials. The following measures should be considered in addressing this level of viral risk remediation:
• Implement a process of identification and segregation of all critical (e.g., animal and human origin) raw materials.
• Introduce a risk assessment for animal- and human-origin components.
• Develop a policy of “three Rs” (replacement/reduction/refinement) for animal- and human-origin components.
• Maintain solid knowledge of raw materials origin, sourcing, manufacturing, testing, storage, and traceability.
• Review of critical raw material specifications for adequacy and viral safety acceptance criteria.
• Maintain a detailed supplier auditing and qualification program that includes biosafety considerations.
• Establish a supplier development and improvement program addressing key areas of quality, biosafety, and risk management.
• Treat raw materials (e.g., through ultraviolent [UV-C] irradiation, gamma irradiation, heat, pH, solvent detergent, nanofiltration, etc.) to clear viruses. Although treatment options are helpful mitigation tools, they are not equally effective against all viruses. From that perspective, raw material treatment provides a certain level of risk mitigation, but not complete risk elimination.
Some examples of raw material (animal origin and chemically defined) treatment options include:
• When serum is used in manufacture, serum treatment by gamma irradiation (30–50 KGy), UV-C (30–150 mJ/cm2), or other modalities may be considered to be practical risk mitigation tools.
• When porcine trypsin is used in manufacturing processes, replacement with recombinant trypsin can be investigated. Alternatively, liquid porcine trypsin solution could be nanofiltered using 15–20-nm pore size filters (circo- and parvovirus removal) or treated by UV-C rays.
• Bulk powder material of animal/human origin (e.g., serum albumin, transferrin) can be gamma irradiated in its final packaging. Alternatively (or additionally), nanofiltration (20-nm pore size) of liquid solutions can be considered at the point of use.
• Liquid cell-culture media can be treated by nanofiltration (20-nm pore size), UV-C, or heat (e.g., high-temperature for short-time treatment, HTST) at the point of use. Gamma irradiation of the media powder before reconstitution may also be investigated realizing that certain media components may be radiosensitive and thus not compatible with gamma irradiation.
• Other raw materials including formulation buffers can be nanofiltered (20-nm pore size) as liquid solutions.
Building viral safety into a quality system
Effective virus risk mitigation should be part of the overall quality system. That way, the viral safety is “system-driven” instead of being people-dependent. Prevention of virus introduction, viral risk understanding/mitigation, and effective response to potential viral contamination are the main objectives of this step. When building viral safety into the overall quality system, a written, comprehensive virus mitigation program is necessary. The main purpose of this program is to promulgate a sustainable, long-term policy as a foundation for viral safety based on the SbD principles. It will form the basis for incorporation of viral safety into the overall quality system for both commercial processes and new products in development.
Additionally, a viral risk assessment must be performed. The purpose of a viral risk assessment is to promote ongoing and proactive viral risk identification and management. It should be conducted using a risk analysis tool suitable for viral risk (e.g., failure mode and effect analysis [FMEA], preliminary hazard analysis [PHA], and risk ranking and filtering [RRF]) with periodic risk re-evaluation, addressing the following areas at minimum:
• Risk of virus entry with appropriate controls:
Starting materials (e.g., cell banks, viral and bacterial seeds, animals used in production)
Raw materials (e.g., cell-culture media, serum, plant extracts)
Personnel
Equipment
Manufacturing process (e.g., type of cells, type of process, open vs. closed cell-culture steps, duration, containment)
Manufacturing plant internal environment and utilities
Outside plant environment
• Specific virus controls in the product manufacturing process:
Virus testing (bulk harvest, drug substance, drug product)
Viral clearance affored by downstream purification.
A written emergency (contamination) response plan is an important part of the SbD method. The objective of a viral response plan is to specify necessary steps in the response process, delineate clear roles and responsibilities, and ensure rapid response to a suspect or confirmed viral contamination. It ought to be specific in terms of clearly addressing the questions of what, why, who, how, when, and where. A successful response plan achieves effective area containment, allows rapid virus elimination through effective disinfection, and enables speedy facility return to the routine manufacturing regimen.
Existing procedures may need to be modified to incorporate viral safety. Certain quality procedures may need to be refined to integrate elements of viral safety, as appropriate. Such procedures may include aseptic training, purchasing of suitable raw materials, raw material supply-chain management, and cleaning and sanitization, for example.
Biosafety should be incorporated into a company’s quality audit program. Incorporation of viral safety elements into the internal and external (supplier) audits helps identify weaknesses and strengths of the firm’s quality systems. Importantly, it helps drive improvements in the practices of the suppliers of critical raw materials.
Lastly, a training module on viral safety and its impact on product and patients would be prudent. This training brings awareness and drives employee behavior in specific units of operations that are more susceptible to viral contamination. In concert, these measures would greatly enhance a quality-based viral risk mitigation program and a firm’s readiness to respond to a contamination event.
Product development
New product development presents an ideal opportunity to incorporate all the relevant principles of SbD into the new process. Here, product safety is intentionally designed into the new process with the goal of preventing introduction of adventitious viruses into the process and designing meaningful product testing strategy, while enabling rapid detection and containment in any area where a problem has occurred. The following points should be considered when building SbD into new product/process development:
• Selection and engineering of cell lines
• Development of animal-origin-free/chemically defined cell banks (e.g., from transfection to master and working cell bank [MCB, WCB] generation)
• Development of animal-origin-free/chemically defined manufacturing processes, devoid of animal and human origin components
• Incorporation of an appropriate and meaningful testing strategy
• Use of upstream viral barrier technologies (e.g., UV-C, HTST, nanofiltration) for media/raw material treatment
• Use of closed process systems where appropriate
• Incorporation of effective, validated viral clearance steps in downstream processing including two orthogonal viral clearance steps (e.g., an inactivation step and a viral removal by nanofiltration) for drug-substance generation
• Design of closed processes/units of operation, making them inaccessible to environmental adventitious agents
• Use of disposable, single-use equipment whenever feasible
• Exploitation of a clear sampling plan and well-defined testing plan for adventitious agents
• Inclusion of process analytical technology (PAT) to enable early detection of cell culture contamination
• Implementation of all the raw materials and quality system principles discussed in the previous sections.
Commercial product manufacturing
Control measures at this level serve primarily to prevent virus introduction and to ensure virus containment. The controls employed at this level may include:
• Continuous process improvement, considering some of the elements discussed for new product development
• Effective facility and equipment cleaning and sanitization procedures using proven virucidal and sporicidal chemicals
• An ongoing facility improvement plan and proper facility design to prevent contamination, including area containment to prevent virus spread from the affected area; segregation of various activities such as raw material handling, media and buffer preparation, cell-culture operations, downstream operations, and post-viral clearance operations; air pressure differential; HVAC; treatment of production water; control and appropriate filtration of production gases; waste treatment and waste disposal; and a pest control program
• Proper employee training and gowning, including policy on managing employees with apparent communicable (respiratory, gastrointestinal, cutaneous) diseases in GMP areas.
• Testing design should be suitable for the process and, at a minimum, in compliance with the current regulatory requirements.
• Testing should be conducted at the most meaningful process steps using appropriate samples, sample volumes, and suitable testing methods.
• Limitations of the existing testing methods used for raw materials, cell banks, seeds, harvests, and product testing should be understood and addressed appropriately.
• Risk-based testing augmentation: specific virus testing (e.g., by polymerase chain reaction [PCR] or other technology) should be implemented, if justified by risk assessment. This testing should include new and emerging agents.
• New detection technology (13) with broad detection and identification capability (e.g., deep sequencing, microarrays) should be adopted to augment the overall testing and characterization program, to address certain testing gaps, or to support risk assessment program.
As the name indicates, the main purpose of this level is to detect adventitious viruses. Early detection is essential in order to deploy an adequate response to contain and eliminate the virus. Various testing methods are used at several stages of process/product development and manufacture. The following testing principles should be considered:
Summary of various virus risk-mitigation options
Although the safety triangle plays a central role in ensuring viral safety of the final product, various other options (Figure 3) are available to further augment the safety tripod and reduce the overall risk of viral entry into the product stream. When properly integrated, these measures can bring residual viral safety risk to a very low level.
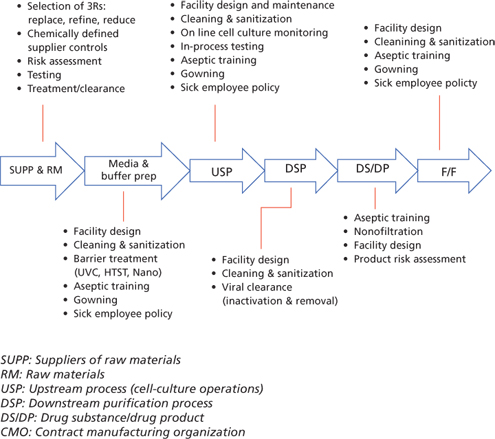
Conclusions
Viral safety is an important quality attribute of a drug product. Endogenous or adventitious viral agents are generally regarded as product impurities and are not acceptable in the final drug dosage. Ensuring freedom from adventitious or endogenous viral agents is therefore crucial for the safety of biological drug recipients. Traditionally, the safety triangle (selection-testing-viral clearance) has been helpful, but it may not be sufficient to meet today’s expectations. An integrated viral risk-mitigation program (i.e., safety by design) across the supply chain, therefore, is important to provide a high level of viral safety of biological products. Safety by design should not only be incorporated into new product development, it should also be part of a larger process-control strategy for both development and commercial products. Zero risk in the manufacture of biological products is not achievable; aiming for risk that is “as low as reasonably achievable” (ALARA) should be an acceptable goal—and it can be achieved through use of a holistic, integrated, and product-specific safety by design program.
References:
1. EMA, Guideline on the Use of Bovine Serum in the Manufacture of Human Biological Medicinal Products (London, May 30, 2013).
2. EMA, Requirements and Controls Applied to Bovine Serum Used in the Production of Immunological Veterinary Medicinal Products (London, Nov. 9, 2009 [Revised]).
3. EMA, Guideline on the Use of Porcine Trypsin Used in the Manufacture of Human Biological Medicinal Products (London, Feb. 20, 2014).
4. EMA, Note for Guidance on Minimising the Risk of Transmitting Animal Spongiform Encephalopathy Agents via Human and Veterinary Medicinal Products (London, 5.3.2011).
5. OIE Terrestrial Animal Health Code, “Bovine spongiform encephalopathy,” (Chapter 11.5, 2011), accessed on March 1, 2016.
6. CFR Title 9, parts 113.46, 113.47, 113.50, 113.51, 113.52, 113.52, 113.54, 113.55 (Government Printing Office, Washington, DC, Jan. 1, 2006 edition), pp. 640–645.
7. USP, <1024> Bovine Serum Appendix 1, USP 38–NF 33 (US Pharmacopeial Convention, Rockville, MD, Oct. 1, 2015), pp. 719.
8. European Pharmacopoeia, Monograph: Bovine Serum, 01/2008:2262, pp. 1506–1507.
9. ICH, Q5A (R1) Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin, Step 5 version (1997).
10. FDA, Memorandum: Points to Consider in Characterization of Cell Lines Used to Produce Biologics (Rockville, MD, Jul. 12, 1993).
11. FDA, Memorandum: Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use (Rockville, MD, Feb. 28, 1997).
12. ICH, Q9 Quality Risk Management (Rockville, MD, January 2011).
13. PDA, Technical Report #71: Emerging Methods for Virus Detection. PDA 2015.
14. ICH, Q5D Quality of Biotechnological Products: Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products (March 1998).
15. World Health Organization (WHO), Recommendations for the Evaluation of Animal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks (2010).
16. EMA, Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products (London, July 24, 2008).
17. R.L. Garnick, Dev. Biol. Stand. 88, pp. 199–203 (1996).
18. H. Rabenau et al., Biologicals 21, pp. 207–214 (1993).
19. R. Nims et al., BioPharm Int. 21 (10), pp. 89–94 (2008).
20. A. Oehmig, J. Gen. Vir. 84 (12), pp. 2837–2845 (2003).
21. A. Kerr and R. Nims, PDA J. Pharm. Sci. Tech. 64, pp. 481–485 (2010).
22. M. Plavsic et al., Bioprocess. J. 9 (2), pp. 6–12, (2011).
23. Y. Qiu et al., Biotechnol. Bioeng.110 (5), pp. 1342–1351 (2013).
24. P.P. Pastoret, Biologicals 38, pp. 332–334 (2010).
25. J.G. Victoria et al., J. Vir. 84 (12), pp. 6033–40 (2010).
26. Pharma IQ, "62.5% of Biologics Professionals Have Strategy in Place to Mitigate Risks of Contamination," accessed June 2, 2015.
About the Author
Mark Plavsic, PhD, DVM, was head of Corporate Product Biosafety, Genzyme, a Sanofi Company, Framingham, MA, USA. Presently, Mark is head of process development and manufacturing with Torque Therapeutics, mark@torque.email.