Jun 01, 2015
By Fengqiang Wang, PhD, Daisy Richardson, Mohammed Shameem
BioPharm International
Volume 28, Issue 6, pg 32–38
 Host-cell proteins (HCPs) constitute a major part of process-related impurities during biologics production. The amount of residual HCPs in drug product is generally considered a critical quality attribute (CQA), due to their potential to affect product safety and efficacy. Therefore, it is a regulatory requirement to monitor the removal of HCPs in drug product during bioprocess development.
Host-cell proteins (HCPs) constitute a major part of process-related impurities during biologics production. The amount of residual HCPs in drug product is generally considered a critical quality attribute (CQA), due to their potential to affect product safety and efficacy. Therefore, it is a regulatory requirement to monitor the removal of HCPs in drug product during bioprocess development.
HCPs are proteins produced or encoded by the host organisms used to produce recombinant therapeutic proteins (1). Recombinant therapeutic proteins are usually produced by genetically-modified prokaryotic or eukaryotic host cells using cell culture/fermentation technology (2). Genetic engineering allows the host cells to be transformed to produce a protein of interest selectively. During the recombinant protein production, host cells also coproduce proteins related to the normal cell functions such as cell growth, proliferation, survival, gene transcription, protein synthesis, and etc. Other non-essential proteins may also be released to the cell culture/fermentation as a result of cell apoptosis/death/lysis. In general, apart from the therapeutic protein of interest, all endogenous proteins co-expressed by the host cells are called host-cell proteins (2).
Risks Associated with HCPs
HCPs constitute a major group of process-related impurities in a drug product. The risks associated with HCPs are primarily immunogenicity. HCPs are complex mixtures with diverse physiochemical and immunological properties (2). Almost all HCPs carry clinical safety risks as foreign proteins due to the potential to elicit immune response in humans. In addition, some HCPs can also act as adjuvants to enhance immune response to a drug product (1, 3). Certain HCPs with proteolytic activity can also affect drug product stability and efficacy if not adequately removed or inactivated (4). HCPs have the potential to affect both the safety and efficacy aspects of a given drug product.
The risks associated with HCPs are often assessed by a combination of downstream process capabilities, residual HCPs levels, the (maximum) dose, route of administration, dosing frequency, toxicological data, and clinical data (1). Although it is a common understanding that HCPs pose clinical safety risk due to their potential to elicit an immune response, it is difficult to demonstrate which HCP and in what concentration may cause immunogenicity problems in humans (1). Theoretically, preclinical pharmacological and toxicological evaluations can be performed with the presence of different amounts of HCP impurities; however, the evaluation results are mostly irrelevant because the magnitude and nature of the immune response depends on the homology of the amino acid sequence, residual HCPs amount, and product dosing regimen. For this reason, a risk control strategy is applied through the development of robust downstream bioprocess to remove HCPs to as low a evel as possible or to “undetectable” levels in drug substance/product (1, 3, 5). The detectability of residual HCPs, however, also depends on the method of detection’s sensitivity. Industry addresses this risk by meticulous method development and the use of multiple technologies to evaluate all potential HCPs that might coproduce or copurify with drug product during bioprocess development.
Regulatory Requirements for HCPs Measurement and Control
According to International Conference on Harmonization (ICH) guidelines Q6B, “For host-cell proteins, a sensitive assay (e.g., immunoassay, capable of detecting a wide range of protein impurities) is generally utilized. In the case of an immunoassay, a polyclonal antibody (pAb) used in the test is generated by immunization with a preparation of a production cell minus the product-coding gene, fusion partners, or other appropriate cell lines” … “Clearance studies, which could include spiking experiments at the laboratory scale, to demonstrate the removal of cell substrate-derived impurities such as nucleic acids and host cell proteins may sometimes be used to eliminate the need for establishing acceptance criteria for these impurities” (6). FDA expects “Whenever possible, contaminants introduced by the recovery and purification process should be below detectable levels using a highly sensitive analytical method” (7). The European Medicines Agency (EMA) guideline CPMP/BWP/382/97 states, “In summary, for HCP, whatever the product and production system, residual HCP have to be tested for on a routine basis”…“As such, it is currently required that HCP be routinely monitored at the purified bulk level, using suitable analytical assays. Results from batch to batch should be consistent and meet specification limits” (8).
Regulatory agencies from other countries and emerging markets may have their own wording on HCP control, but it is generally accepted that a sensitive, validated method is required to monitor residual HCPs in accordance with ICH guidelines. The allowed amount of residual HCPs in final bulk material is determined on a case-by-case basis, commonly in the 1 to 100 ng/mg range.
Measuring and Monitoring Residual HCPs
To date, immunoassay, commonly in the form of sandwich enzyme-linked immunosorbent assay (ELISA) (see Figure 1), remains as the industry gold standard for HCP measurement due to its high sensitivity and high throughput (3, 5). The HCP composition and abundance are unique to their respective host and the manufacturing process used for biologics production. Meanwhile, different host cells and manufacturing processes may produce certain HCPs in similar abundance. The number of proteins derived from host cells varies significantly from host to host. For example, Escherichia coli (E. coli) has ~4300 genes, whereas Chinese Hamster Ovary (CHO) cells have ~30,000 genes (1). Although not every host gene will be transcribed and translated to protein, the complexity of host genome and the post-translational modification present in mammalian cells make it almost impossible to understand the complete HCP composition in a given manufacturing process. Because these HCPs are potentially immunogenic, a commonly accepted method to evaluate the presence of HCPs is through an immunoassay. In theory, an HCP mixture injected into an animal, such as a rabbit, goat, or chicken, will elicit an immune response, and the animals will generate anti-HCP antibodies against these foreign proteins. Although the identities of all HCPs are not known, the polyclonal antibodies raised in animals should be able to recognize most, if not all, of the proteins contained in the HCP mixture. Using these polyclonal antibodies, a multi-analyte sandwich ELISA can be developed (see Figure 1). In Figure 1, capture antibodies enrich the HCPs from sample and immobilize them to a 96 (or greater)-well plate. Then, detection antibodies, conjugated directly with an enzyme or through a biotin-avidin magnification, bind to the captured HCPs. An enzyme, commonly horseradish peroxidase (HRP), can catalyze the substrate to generate a colorimetric, chemiluminescent, or fluorescent signal that correlates with the amount of HCPs in the test sample (5).
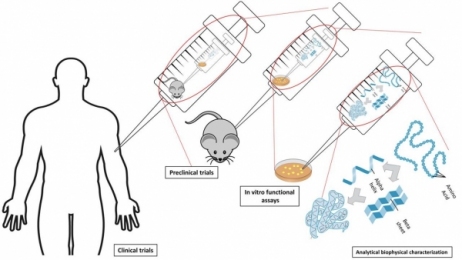 Figure 1: Schematic view of sandwich enzyme-linked immunosorbent assay (ELISA) used for host-cell protein (HCP) detection and measurement.
Figure 1: Schematic view of sandwich enzyme-linked immunosorbent assay (ELISA) used for host-cell protein (HCP) detection and measurement.
Generic HCP ELISA developed using polyclonal antibodies raised against parental cell lysate or cell culture supernatant allows the detection of a majority of HCP species, but may not be able to detect a subgroup of proteins specific to a certain manufacturing process. A generic ELISA kit is commercially available and has the advantage of eliminating lengthy assay development time as indicated in Figure 2, with only steps pointed in yellow being required before its use for process development. This makes the kit good for early phase development. In late phase (Phase III or commercial), cell line-specific platform assay or upstream process-specific ELISA assay is often required to mitigate the risk associated with a more generic commercial assay. The development cycle for an in-house HCP assay is shown in Figure 2 following the flow of green arrows. Commonly, platform- or process-specific HCPs are generated by growing a null cell culture (mock) without the product-encoding gene under similar upstream processes to represent HCPs from a (platform) production culture. PAbs generated by immunizing animals with these HCPs will be used for ELISA development upon qualification by a 2D-coverage assessment. The pAbs should recognize a large majority of HCPs coproduced with drug product. When less-than-ideal coverage is observed, different strategies (as represented by the orange arrows) can be used to improve the coverage or qualify the ELISA at risk and supplement the ELISA with orthogonal methods to characterize HCP process clearance (see Figure 2). Upon qualification and validation, HCP ELISA can serve as a QC release assay for drug substance, and in-process pool testing results can guide the downstream process development (see Figure 2).
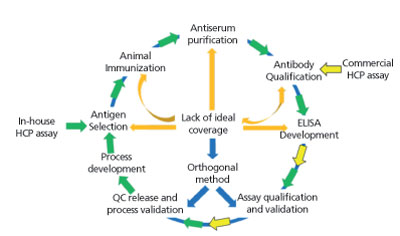 Figure 2: Schematic view of host-cell protein (HCP) assay development cycle.
Figure 2: Schematic view of host-cell protein (HCP) assay development cycle.
In a typical recovery process for a therapeutic monoclonal antibody, the highest concentration of HCP is detected in the harvested cell culture fluid (HCCF), and then cleared through additional downstream purification steps, with typically low (1-100 ng/mg, or ppm) levels of HCPs observed in final bulk drug substance (5).
Limitations of HCP ELISA
Due to the heterogeneity of HCPs, proteins of high abundance/immunogenicity often dominate the multi-analyte ELISA signals, whereas proteins of low abundance/immunogenicity don’t have enough antibodies to recognize them. In addition, copurifying HCPs enriched in drug substance may also have limited antibody to detect them, resulting in dilutional nonlinearity and potentially an underestimate of residual HCPs. Furthermore, due to the fact that not every HCP is immunogenic in animals, even the best process-specific HCP ELISA cannot detect 100% of HCPs coproduced with the recombinant protein. Supplementary to HCP ELISA, traditional HCP separation and visualization methods, such as 1D and 2D sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), remain useful tools for HCP characterization. Other technologies, such as 2D-differential in-gel electrophoresis (DIGE), capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry (CZE-ESI-MS/MS) or two-dimensional-liquid chromatography-tandem mass spectrometry (2D-LC-MS/MS) also emerged as orthogonal methods for HCP characterization (1, 2, 9-12).
Zhu-Shimoni et al from Genentech published a comprehensive review on the limitations of HCP testing by ELISA with some examples from their process development (5). One example is a HCP named GST-α. GST-α causes no or low immune response in the animals being immunized and thus doesn’t generate an antibody in the polyclonal antibodies used for HCP ELISA. However, this protein was detected on capillary electrophoresis-sodium dodecyl sulfate (CE-SDS) as an impurity peak with a ~2000 ng/mg relative concentration in bulk drug substance. Additionally, some of HCPs may copurify with recombinant protein and thus become enriched in the final bulk; however, the antibodies against these HCPs in the polyclonal antiserum raised against a mock HCP pool may have limited quantity to accurately detect them. Furthermore, steric hindrance and the lack of multiple epitopes to bind the same antibody (capture and detection) can cause inaccuracy on HCP measurement (5). The lack of appropriate calibration standards also limits the accuracy of HCP quantification. These limitations of HCP ELISA mentioned previously can be overcome using orthogonal methods.
A large amount (> 0.1%) of residual HCPs present in final drug substance can be detected by size exclusion (SEC), ion exchange (IEX), or reversed phase (RP)-high performance liquid chromatography (HPLC), SDS-PAGE, CE-SDS, isolectric focusing capillary electrophoresis (iCE), and other analytical methods and confirmed with LC-MS. However, most HCPs are present in the final drug substance at very low levels and require much more sensitive method for detection. Analytical methods mentioned previously often don’t have the sensitivity and resolution to separate and detect individual HCP. Immunoassay-based methods, such as slot blot assay and western blot assay, have the sensitivity but often are only semi-quantitative (1, 3, 13). Protein separation and visualization methods, such as 2D-SDS-PAGE, supplement HCP ELISA by providing information on individual HCP properties. 2D-SDS-PAGE with silver stain or other sensitive staining method, such as Sypro Ruby (Life Technologies, Grand Island, NY), has traditionally been used to analyze complex protein mixtures due to its ability to separate proteins by both isoelectric point (pI) and molecular weight (MW). Silver stain has a detection sensitivity of ~0.2-0.5 ng/protein spot, thus can detect protein of very low abundance, but has a lengthy and tedious staining procedure as well as gel-to-gel variations. To overcome the gel-to-gel variation, two or three samples can be run on the same gel using DIGE, with each of the samples pre-labeled with different fluorescent dyes (2). Analyzing drug substance or in-process pools with 2D-DIGE can give the laboratory scientist a snapshot of the HCP profile, and an easy comparison of the HCP composition differences between two samples (see Figure 3). Coupled with spot picking and LC-MS, 2D-DIGE provides direct information on the HCP properties (pI, MW, abundance, and identification), which can help improve HCP removal strategy in downstream process development.
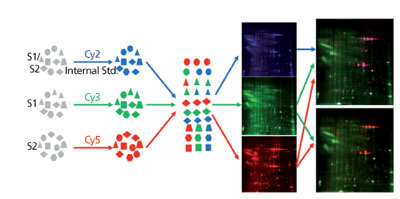 Figure 3: An example on the use of 2D-differential in-gel electrophoresis for hostcell protein profile comparison between a null cell culture (mock, green) and a monoclonal antibody production harvest cell culture fluid (HCCF, red).
Figure 3: An example on the use of 2D-differential in-gel electrophoresis for hostcell protein profile comparison between a null cell culture (mock, green) and a monoclonal antibody production harvest cell culture fluid (HCCF, red).
In the past five years, 2D-LC-MS has emerged as a new orthogonal method for HCP characterization, with the advantage of being sensitive, specific, gel-free and automatable. With an additional separation dimension by high pH reversed phase, ion-exchange, or size-exclusion before LC-MS/MS identification, 2D-LC-MS reduces the interference from dominant peptides digested from recombinant protein and maximizes the detection of low abundance HCP-related peptides (9, 11, 14, 15). Additionally, with the 2D-LC-MS identification of potential residual HCPs, a targeted multiple reaction monitoring (MRM)-LC-MS-based method can be developed to quantify multiple HCPs in a drug substance (5, 11, 16, 17).
Summary
Adequate HCP removal is an important indication of robust and well-controlled bioprocessing. Due to the generally low abundance of HCPs in drug substance, however, it is quite a challenge to detect and measure HCPs in a matrix dominated by the recombinant protein. HCP ELISA has the sensitivity to detect ng/mg level of residual HCPs in drug substance but is limited by the pAbs used in the assay. Often, the pAbs are raised against an HCP population present in the upstream of the bioprocess or a more generic host-cell proteome. These antibodies are not specific enough to the downstream process and thus, the amount of available antibodies present are not correlated with the amount of HCPs in drug substance, which results in antigen excess and lack of dilutional linearity. On the other hand, downstream process-specific ELISA can miss certain HCPs that leak through the particular downstream purification process and/or when downstream processes change. Developing a process-specific HCP ELISA for each biologic is also costly and time-consuming, given the time required to generate process-specific reagents, antibodies, and to develop and validate the assay. For this reason, a multi-product platform assay is more feasible and cost-effective if using the same cell line or cell lines with similar HCP profiles. Data indicate that different upstream processes only change the expression/secretion of a very small subgroup of HCPs within thousands of proteins (18). Using either platform HCP ELISA or process-specific HCP ELISA for HCP testing and control is a strategic decision balancing the benefit and risk of each. No matter which process is chosen, careful qualification of the antibody reagents used in ELISA for each program is necessary to demonstrate their coverage of the large majority of HCPs potentially coproduced with a therapeutic protein. In addition, orthogonal methods such as 2D-DIGE and 2D-LC-MS can be used to provide additional evidence on the process robustness in HCP clearance. These methods are also able to characterize HCPs copurified with recombinant protein that do not elicit an immune response in animals.
When used together with HCP ELISA, the aforementioned characterization methods reduce the risk of HCP oversight and offer valuable information on the HCP properties and identities to guide downstream process development. Furthermore, single- or multi-analyte ELISA can be developed to target copurified HCPs upon LC-MS/MS identification and the understanding of antigen excess in platform- or process-specific HCP ELISA.
Acknowledgements
The authors would like to thank Drs. Steve Farrand, David Chin, and Zhi Chen for their thorough review of this manuscript.
References
1. X. Wang, A.K. Hunter, and N.M. Mozier, Biotechnol. Bioeng. 103 (3), pp. 446-58, 2009.
2. M. Jin, et al., Biotechnol. Bioeng. 105 (2), pp. 306-16, 2010.
3. L.C. Eaton, J. Chromatogr. A 705 (1), pp. 105-14, 1995.
4. F. Robert, et al., Biotechnol. Bioeng. 104 (6), pp. 1132-41, 2009.
5. J. Zhu-Shimoni, et al., Biotechnol Bioeng. 111 (12), pp. 2367-79, 2014.
6. ICH, Q6B, Specifications: Test Procedures And Acceptance Criteria For Biotechnological/Biological Products (ICH, March 10, 1999), www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf
7. FDA, Points to Consider in the Manufacture & Testing of Monoclonal Products for Human Use (1997), https://www.fda.gov/media/76798/download
8. The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit, London, June 10, 1997, CPMP/BWP/382/97, www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003322.pdf
9. G. Zhu, et al., Electrophoresis 35 (10), pp. 1448-52, 2014.
10. A. L. Tscheliessnig, J. Konrath, R. Bates, et al., Biotechnol. J. 8 (6), pp. 655-70, 2013.
11. M.R. Schenauer, G.C. Flynn, and A.M. Goetze, Anal. Biochem. 428 (2), pp. 150-7, 2012.
12. C.E. Hogwood, D.G. Bracewell, and C.M. Smales, Curr. Opin. Biotechnol. 30C, pp. 153-160, 2014
13. D. Zhu, A.J. Saul, and A.P. Miles, J. Immunol. Methods, 306 (1-2), pp. 40-50, 2005.
14. M.R. Schenauer, G.C. Flynn, and A.M. Goetze, Biotechnol. Prog. 29 (4), pp. 951-7, 2013.
15. J. H. Thompson, et al., Rapid Commun. Mass Spectrom. 28 (8), pp. 855-60, 2014.
16. C. E. Doneanu and W. Chen, Methods Mol. Biol. 1129, pp. 341-50, 2014.
18. D.C. Krawitz, et al., Proteomics, 6 (1), pp. 94-110, 2006.
Article Details
BioPharm International
Vol. 28, No. 6
Pages: 32–38
Citation: When referring to this article, please cite it as F. Wang, D. Richardson, and M. Shameem, “Host-Cell Protein Measurement and Control” BioPharm International 28 (6) 2015.