Modeling the Degradation of mAb Therapeutics
By Anurag S. Rathore, Rohit Bansal
 sakurra/Stock.Adobe.com
sakurra/Stock.Adobe.com
Ensuring the stability of biotherapeutic products, which has an impact on their safety and efficacy, continues to be a major challenge for the biopharmaceutical industry. This is particularly true for monoclonal antibodies (mAbs), which account for most commercial biopharmaceuticals today. Both physical effects such as aggregation and chemical modifications (e.g., fragmentation) are the two leading paths for mAb degradation. Kinetic modeling can be an effective tool for establishing product stability, offering a deeper understanding of these processes and allowing developers to mitigate the risk of product degradation during the early stages of process and product development.
Proteins pose stability risks
Monoclonal antibodies are currently the leading class of protein therapeutic molecules due to their ability to treat a wide range of lethal and life-threatening diseases, including cancer, Ebola (1, 2), multiple sclerosis, rheumatoid arthritis, psoriasis, and asthma. FDA has approved a number of these antibody drug molecules, which are in various stages of clinical and pre-clinical development. But since the major constituent of the final drug product is protein, there is a significant risk associated with the stability of these drug molecules (3–5).
Instability associated with various critical quality attributes (e.g., aggregation, fragmentation, charge variants, and glycosylation) continues to be a major concern for the biopharmaceutical industry. It can, either directly or indirectly, affect mAbs’ biological activity as well as toxicity (4).
The instabilities associated with mAbs can be broadly classified as either physical or chemical (3, 4). Aggregation is one of the major physical instabilities that is believed to impact the immunogenicity of a therapeutic (5).
Different types of chemical instabilities include deamidation, oxidation, fragmentation, and hydrolysis. Although the effects of these instabilities (both physical as well as chemical) may differ among various classes of mAb molecules, biopharmaceutical manufacturers desire to minimize them (2). Aggregation and fragmentation are the two most significant degradation pathways for mAbs (6–7). They result in instabilities that can occur at various stages of product development, including upstream processing (during protein expression in cell culture), downstream processing (purification), product formulation, storage, and transport (6, 7).
Factors that affect mAb stability
Factors that affect the stability of an mAb therapeutic can relate either to its structure (primary, secondary, or tertiary), or to its environment. In the primary structure, a protein is more susceptible to instability if there is change in its surface charge distribution or hydrophobicity. Alterations in the secondary structure, especially an increase in beta content of the protein molecule, can make it significantly more vulnerable to aggregation. Environmental factors that can have an impact on the protein include pH, temperature, salt concentration, buffer type, protein concentration, ionic strength, mixing, shear, metal ions, pressure, freeze thawing, freeze drying, and reconstitution (4, 8).
A protein may change in response to these factors, altering its physical structure either by physical association (aggregation) or chemical degradation (fragmentation). This article in the Elements of Biopharmaceutical Production discusses the kinetics of these degradation pathways as well as the insights they reveal that can help developers ensure the stability of biotherapeutic products.
Case study I: aggregation of mAb products
Modeling the kinetics and thermodynamics of protein aggregation can prove valuable in understanding its mechanism (9). Mathematical modeling of protein aggregation correlated along with kinetic data can help developers gain both qualitative and quantitative insights into the mechanism behind aggregation. Armed with this knowledge, developers can better predict and control aggregation by optimizing timeframes and environmental conditions (10).
A number of protein aggregation models already exist (9), including the Lumry-Eyring model, which is one of the most commonly used models to predict aggregation kinetics. According to this model, aggregation occurs in two steps: conformational reversible unfolding of the protein molecule followed by irreversible assembly into aggregates, which are either physically or chemically linked to each other.
Many researchers have modified the Lumry Eyring model to account for nucleation as well as higher order aggregation (10). Examples are the Extended Lumry Eyring (ELE) Model and the Lumry Eyring Nucleated Polymerization (LENP) Model (11). In addition, alternative models (9) have proposed alternate aggregation models for proteins such as prions (12), amyloid β (13), and insulin (14).
Lumry Eyring Nucleated Polymerization (LENP)
The Lumry Eyring model introduces the concept of nucleation to aggregation. The kinetic reaction scheme presented in this model depends on:
- • Order of reaction
- • Starting protein concentration
- • Size distribution amongst different aggregate species (6).
The parameters which are considered in this model are nucleus stoichiometry (x), monomers added in each growth step (δ), and the inverse rate coefficients for nucleation and growth (10). These rate coefficients denote the corresponding time scales (τnand τg) and the details of the model can be found in the literature (10).
Extended Lumry Eyring model (ELE)
Another model used for kinetic analysis of aggregation is the extended Lumry-Eyring (ELE) model. With this approach, unfolding is considered as a single reversible rate-limiting reaction with both folded native and reactive unfolded monomer species in thermodynamic equilibrium with each other (11, 15, 16). This model takes into consideration both reversible and irreversible conformational changes that occur during aggregation (17). It also considers the solution’s conformational and kinetic colloidal stability. Concentration-based experimental data are fitted into model equations to calculate the apparent rate constants and predict the monomer degradation rate (17).
Aggregate sample prep and characterization
To monitor and understand the aggregation mechanism and its behavior in proteins, a model protein (mAb) was chosen with an isoelectric point (pI) of 8.5. Buffer exchange was used to monitor the aggregation kinetics of this mAb under the conditions that are prevalent in the biopharmaceutical industry during downstream purification.
Detailed experimental procedures and conditions have been described elsewhere (6). In brief, buffers examined included those commonly used for Protein A chromatography, cation exchange chromatography, and anion exchange chromatography.
Samples were kept at three different temperatures (4 °C, 15 °C, and 30 °C) at a concentration of 10 mg/mL. Sampling was performed over 120 hours at intermittent time points. The samples were analyzed for aggregation content using size-exclusion high performance liquid chromatography (SE–HPLC) and types of oligomers using dynamic light scattering (DLS) (7) (Figure 1A).
All the experiments were done in triplicate to ensure reproducibility. Aggregation data obtained by SE–HPLC were analyzed using MATLAB R2011a and fitted into ELE and LENP models. Sets of ordinary differential equations (ODEs) were then solved using Gauss-Newtonian algorithm to estimate the model parameters (18).
Factors affecting mAb aggregation
Aggregation was found to be at its maximum at low pH and accelerated upon increasing temperature and salt concentration (9). Aggregation at low pH has been primarily associated with changes in the fragment crystallizable (Fc) domain of the antibody (19). These changes include partial unfolding of the Fc domain of the mAb, which leads to the exposure of the hydrophobic residues. These hydrophobic residues, which were previously buried inside the molecule, became attracted to each other. This resulted in the enhancement of aggregation at low pH. Figure 1B illustrates that aggregation is minimal in cation and anion exchange (CEX and AEX) buffer conditions (high pH) as compared to protein A purification conditions (low pH). Further, higher temperature resulted in partial or complete unfolding of mAb structures and resulted in aggregation due to instability of the protein molecule at higher temperature (Figure 1B).
Presence of salt also affects protein stability and the extent depends on the type of salt, its concentration, protein charge, and protein and salt interaction (Figure 1B) (6). An increase in the rate of aggregation is seen with increasing salt concentration. Apart from these factors, the type of buffer species also seems to have a profound effect on the stability of protein products.
Higher stability is seen in glycine buffer, as compared with citrate and acetate buffers (Figure 1B) (6), governed by the interaction of the Fc domain of the mAb molecule with different buffer species (20). Results obtained from circular dichroism (CD) spectroscopy and DLS measurements support these observations as well (6). To summarize the key findings, it can be said that pH is the most important factor in protein aggregation, followed by temperature, salt concentration, and buffer species.
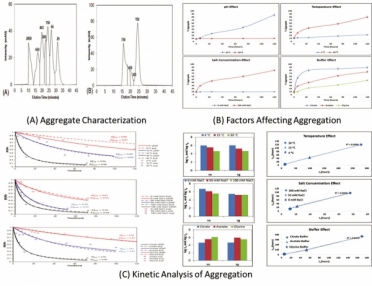
Figure 1: Aggregation: characterization, analysis, and modeling data
[All figures courtesy of the authors].
Kinetic modeling of mAb aggregation
Data obtained from SE–HPLC analysis were fitted using both ELE and LENP models, and the accuracy of the models was judged by doing a comparative evaluation of the experimental values with the model predicted values. Curve fitting for computing the coefficient of regression (R2value) was performed using both models. A comparison of the models found that that the LENP model did a better job of predicting the aggregation kinetics than ELE (6).
Results were then used to compute the time scales of nucleation (τn) and growth (τg) in order to predict aggregation kinetics (Figure 1C). The modeling parameters of nucleation and growth time scales were compared across different incubation conditions. It was noted that, as the temperature is increased from 4 ºC to 30 ºC (in the case of citrate buffer at pH 3.0 with 100 mM NaCl), the nucleation and growth time scales decrease, implying faster aggregation (6) (Figure 1C).
There is also a linear relationship between nucleation time scale and monomer half-life as temperature is increased from 4 ºC to 30 ºC. The steeper monomer loss at high temperature is attributed to the increase in the collision frequency due to an increase in protein diffusion.
A similar effect was observed upon increasing the salt concentration from 0 mM NaCl to 100 mM NaCl (Figure 1C). This is likely due to salt ionization, which weakens protein structure and decreases stability because of hydrophobic interactions and weakening of electrostatic repulsions as the salt concentration increases.
Overall, for the system under consideration, the citrate buffer at pH 3.0 showed the steepest monomer loss as compared to acetate and glycine buffers (6) (Figure 1C). There is a linear correlation between nucleation time scale and monomer half-life across the different conditions, with slope between 0.4–0.5, indicating that nucleation dominates aggregation in mAbs and hence that the LENP model offers a better fit than the ELE model (6) (Figure 1C).
Case study II: fragmentation of mAbs
Fragmentation involves the breaking down of the mAb into smaller units, fueled by thermal or chemical energy (21). Fragmentation results in the breakage of bonds between the amino acids, transforming the primary structure of the molecule and, ultimately, other higher order entities. Cleavage of the peptide bond (hydrolysis) at the mAb’s hinge region leads to the formation of Fc -Fab and Fab species (22).
Responses from different classes of the mAb molecules vary with respect to the fragmentation phenomena, depending on each one’s inherited primary structure. Various contaminants (e.g., metal ions and proteases) in the formulation buffer systems significantly affect the extent of fragmentation (22), and degradation can occur at various stages of mAb production and processing (23). Size-based monitoring tools such as SEC, DLS, mass spectrometry (MS), and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) are commonly used to detect fragmentation and identify the various species that are formed (24).
Fragmentation sample prep and characterization
The mAb sample was diafiltered in the formulation buffer (15 mM phosphate, pH 6.5 with 150 mM NaCl, and 0.02% Polysorbate 80) at a final concentration of 5 mg/mL. This sample was then incubated at 50 °C, and the extent of fragmentation was monitored by SE–HPLC at regular time intervals (Figure 2A and 2B).
Various other analytical techniques (e.g., DLS and SDS–PAGE [7]) were used to confirm the sizes of the fragments that were formed (Figure 2D). Reverse-phase high-performance liquid chromatography with mass spectrometry (RPHPLC–MS) was used to analyze the intact mAb samples as well as the fragmented mAb sample and the purified and enriched fragment species to locate the site of fragmentation in the mAb molecule (Figure 2C).
Results of the SEC, DLS, SDS–PAGE, and MS analysis found that two different fragment species were formed upon fragmentation of intact monomer (148 KDa) mAb molecule—Fragment 1 (96.9 KDa) and Fragment 2 (47.3 KDa) (Figures 2C and 2D). This shows that cleavage occurs at the hinge region of the mAb, leading to the formation of Fc–Fab and Fab fragments and corresponding to the published results (7).
Kinetic modeling of mAb fragmentation
Upon observing the fragmentation trend via SEC, it can be inferred that the monomer mAb molecules (M) break down into two species (F1 and F2) with time. Following the temporal trend in concentration change of these three species (M, F1, and F2), it was noticed that, initially, for a period of 72 hours, there is a slow decrease in the concentration of M followed by an abrupt rise in the slope. F1 and F2 fragments also follow this trend in incremental fashion (7) (Figure 2A). This time-based analysis was performed at different temperatures to confirm our hypothesis, upon which a fragmentation model was based. The authors suggest that the phenomenon of fragmentation is autocatalytic in nature based on the assumption that a threshold concentration of fragments is needed to react with intact monomer molecule to further accelerate its degradation (7). To confirm that the proposed model is general in nature, it was tested on another buffer system at 50 °C (15mM phosphate, Ionic strength -29mM, pH 6.8, and 0.015% PS80) (Figure 2B) (22).
After analyzing the experimental results of the two datasets, it was found that, although the fragmentation trend was identical in both the buffer systems, the rate of fragmentation was a bit different because of the different ionic strengths of the two buffers. Approximately 57% of the monomer mAb transformed into fragment species in four days. The kinetic model of fragmentation was developed based on results obtained from SEC. SEC analysis has shown that aggregates’ percentage was constant throughout the study, proving their minimal impact on the fragmentation process.
Sudden increase in fragmentation rate seen
In addition, the fragmentation rate was initially gradual but increased suddenly on the fourth day until the sample had completely degraded. Further, it was observed that the peak next to the monomer peak is the first fragment species (F1 or Fc–Fab), and the second peak is the second fragment species (F2 or Fab).
The concentration of F1 species increases in the beginning and then decreases (7). The concentration of F2 species continuously increases throughout the study. On examining these observations, the following mechanism is proposed:

Moreover, this model was verified experimentally at three different temperatures (40 °C, 45 °C, and 50 °C). The results obtained from kinetic analysis showed that reaction [1] is a non-Arrhenius reaction with negative activation energy. This was also checked at further two temperatures (30 °C, 35 °C). At all the five temperatures, the trend is non-Arrhenius with negative activation energy (Table I).

Table I: Kinetic rate constants obtained using the constructed mechanism for same sample stressed at three different temperatures.
The reaction [3] was confirmed by performing purifying and enriching F1 and F2 species and incubating them at 50 °C separately. F1 species (Fc –Fab) dissociate into Fc and Fab fragment species and F2 fragment was found to be the final end product after which the sample degrades completely (Figure 2E) (7).
Amongst all rate constants, while k1did not follow Arrhenius kinetics, k3and k4did with R2greater than 0.75. Further, k3and k4 exhibited a linear relationship with temperature corresponding to increased fragmentation (Table II).
Table II: R2 value of the fit of linearized Arrhenius plot of ln(k) vs (1/T). The table includes the activation energy values as well as the fit equations.
MATLAB R2015b was used to analyze fragmentation data obtained by SE–HPLC, which was then fitted into the proposed model equations to obtain the rate constants (Figure 2F). For all the fragment species, the R2 value was found to be greater than 0.9, proving that the model is statistically significant (7).
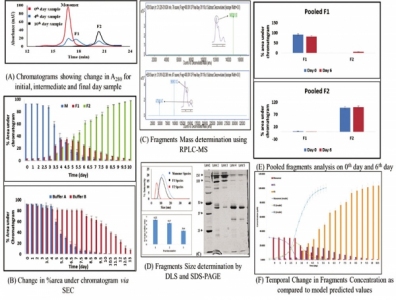
Figure 2: Fragmentation: characterization, analysis, and modeling data.
Summary
Because they are proteins, mAbs are highly sensitive labile molecules and degrade easily via aggregation and fragmentation. This article has discussed kinetic modeling and shown how it can be used as a tool for the early assessment of protein stability.
Insights presented in this article and the accompanying references can help readers in the development of plans for examining and controlling product degradation during production and storage. Such studies open the door for better understanding of mAb degradation pathways, which can only help efforts aimed at increasing the shelf life of mAb drug products.
Acknowledgments
This work was funded by the Center of Excellence for Biopharmaceutical Technology grant from the Indian government’s department of Biotechnology (number BT/COE/34/SP15097/2015).
References
1. N. C. Nicolaides, P. M. Sass, and L. Grasso, Drug Dev Res. 67 (10) 781–789 (2006).
2. M. Vázquez-Rey and D. A. Lang, Biotechnol Bioeng.108 (7) 1494–1508 (2011).
3. T. Ishikawa, T. Ito, R. Endo, et al., Biol. Pharm. Bull.33 (8, 1413-1417 (2010).
4. A. S. Rathore, V. Joshi, and N. Yadav, BioPharm Int.26 (3) 40–45 (2013).
5. I. Arora, R. Bansal, V. Joshi, et al., Intl. J. Chem. Eng. App.5 (5) 433 (2014).
6. A. Singla, R. Bansal, V. Joshi, et al., AAPS J. 18 (3), 689–702 (2016).
7. S. Ravuluri, R. Bansal, N. Chhabra, et al., Pharm. Res.35 (7) 142 (2018).
8. W. Wang et al., Aggregation of Therapeutic Proteins,W. Wang, and C. J. Roberts, Eds. (John Wiley & Sons, Hoboken, NJ, 2010).
9. A. M. Morris, M. A. Watzky, and R. G. Finke, Biochim. Biophys. Acta, Proteins Proteomics. 1794 (3) 375–397 (2009).
10. J. M. Andrews, and C. J. Roberts, J. Phy. Chem. B. 111 (27) 7897–7913 (2007).
11. R. K. Brummitt, D. P. Nesta, L. Chang, et al., J. Pharm. Sci. 100 (6) 2104–2119 (2011).
12. S. B. Prusiner,Science252 (5012) 1515–1522 (1991).
13. A. Lomakin, D. S. Chung, G. B. Benedek, et al., Proc. Natl. Acad. Sci.93 (3) 1125–1129 (1996).
14. T. J. Gibson and R. M. Murphy, Protein Sci., 15(5) 1133–1141 (2006).
15. J. M. Sanchez-Ruiz, Biophy. J.61 (4) 921–935 (1992).
16. P. Arosio, G. Barolo, T. Müller-Späth, H. Wu, et al., Pharm. Res.28 (8) 1884–1894 (2011).
17. L. Nicoud, P. Arosio, M. Sozo, et al., J. Phys. Chem. B. 118 (36) 10595–10606 (2014).
18. C. J. Roberts, J. Phy. Chem. B.107 (5) 1194–1207 (2003).
19. D. Kameoka, E. Masuzaki, T. Ueda, et al., Biochem.142 (3) 383–391 (2007).
20. B. A. Salinas, H. A. Sathish, A. U. Shah, et al., J. Pharm. Sci. 99 (7) 2962–2974 (2010).
21. G. Gaza-Bulseco and H. Liu, Pharm. Res.25 (8) 1881–1890 (2008).
22. S. X. Gao, Y. Zhang, K. Stansberry‐Perkins, et al., Biotechnol. Bioeng. 108 (4) 977–982 (2011).
23. A. J. Cordoba, B. J. Shyong, D. Breen, et al., J Chromatogr B.818 (2) 115–121 (2005).
24. S. Zheng, D. Qiu, M. Adams, et al., AAPS PharmSciTech. 18 (1) 42–48 (2017).
About the Authors
Anurag S. Rathore* is a professor in the department of Chemical Engineering at the Indian Institute of Technology (Delhi) and a member of BioPharm International’s Editorial Advisory Board, asrathore@biotechcmz.com. Rohit Bansal is PhD Research Scholar in the De-partment of Chemical Engineering at the Indian Institute of Technology (Delhi).
*To whom all correspondence should be addressed