Single-domain antibodies are emerging as credible alternatives due to their target specificity, high affinity, and cost-effective recombinant production.
 Smaller recombinant antibody fragments as single-domain antibodies (sdAbs) are emerging as credible alternatives because of their target specificity, high affinity, and cost-effective recombinant production. sdAbs have been forged into multivalent and multispecific therapeutics, or targeting moieties, that are able to shuttle their linked therapeutic cargo (i.e., drugs, nanoparticles, toxins, enzymes, and radionuclides) to the receptor of interest. Their ability to permeate across the blood brain barrier is receiving industrial interest for neurological and neuro-oncological indications.
Smaller recombinant antibody fragments as single-domain antibodies (sdAbs) are emerging as credible alternatives because of their target specificity, high affinity, and cost-effective recombinant production. sdAbs have been forged into multivalent and multispecific therapeutics, or targeting moieties, that are able to shuttle their linked therapeutic cargo (i.e., drugs, nanoparticles, toxins, enzymes, and radionuclides) to the receptor of interest. Their ability to permeate across the blood brain barrier is receiving industrial interest for neurological and neuro-oncological indications.
Going single domain
Biologic therapies such as monoclonal antibodies (mAbs) are enabling the targeted, personalized treatment of cancer, inflammatory diseases, infectious diseases, and diseases of the central nervous system. With 25 mAb products on the market and more than 100 undergoing clinical trials (1-3), it is evident that engineered antibodies have come of age as biopharmaceuticals (4).
Intact antibodies (immunoglobulins IgG, IgM, IgA, and IgE) are highly specific targeting reagents that form part of our key defence against pathogens and toxins. IgG, the main serum antibody, the intact form of which is almost exclusively used in therapeutic antibodies, is a Y-shaped, multidomain protein with antigen-binding sites located on the two Fab (antigen-binding fragment) tips and effector functions mediated by the stem Fc (crystallizable fragment) domain (see Figure 1). Antibodies are bivalent, (i.e., able to bind to two antigens with high affinity and for long retention times [avidity] on many cell-surface receptors and polyvalent antigents). There is, however, a range of applications for which the Fc-mediated effects are neither required nor are desirable. Inappropriate activation of the Fc receptor-expressing cells by antibodies can lead to cytokine release and toxicity. On the other hand, their long circulation half-life results in poor contrast in imaging applications.

In comparison to the integral antibody, the Fab shows an increased ability to penetrate dense tissues as solid tumors and the Fv (variable domain) appears to be even more effective (5). Hence, the reduction of the Fabs to a monomeric domain unit, as VH or VL, should be, theoretically, more effective in the recognition of the associated antigen. Thus, dissecting IgGs initially through enzymatic proteolysis (e.g., with papain or pepsin) and later genetically engineering them into their constituent monovalent domains or bivalent fragments (Fab2, diabodies, and minibodies) (see Figure 2) would improve their clinical value. Smaller recombinant antibody fragments such as monovalent antibody fragments (Fab, scFv) and engineered variants (diabodies, triabodies, minibodies, sdAbs) along with antibody-drug or antibody-nanoparticle conjugates are emerging as credible alternatives. Many of these products are under clinical investigations (4) as Big Pharma and small and medium enterprises (SMEs) continue to make significant investments in bringing these new therapeutics to the market. The main drivers include the more economical production and unique and superior properties for a range of diagnostic and therapeutic applications.

Small, stable, and easy to engineer
In the late 1980s, a sdAb was firstly isolated by Greg Winter’s group at the MRC Laboratory of Molecular Biology in Cambridge, UK, comprising only a VH domain of an antibody (6). It was four times smaller than a Fab and half of the size of Fv. Conventional antibodies are large (150 kDa), while small recombinant antibodies have a mass range between 25-50 kDa. A sdAb is defined as the smallest antigen-binding fragment of antibodies, ranging from 11 to 15 kDa, and consists of a variable domain of an antibody VH or VL chain containing three or six naturally occurring complementary determining regions (CDRs) that permit specific recognition of the antigen (7).
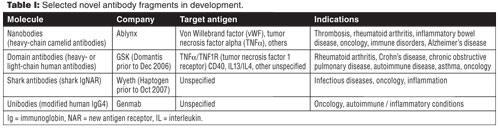
sdAbs have several benefits compared to other antigen-binding units (8). Firstly, sdAbs are antibody-derived fragments that can be expressed in bacteria as active, soluble, and robust proteins. Additionally, single variable domains are known to be able to potentially target cryptic epitopes that are poorly accessible to intact antibodies but enable pathogen to bind to their target receptors (6, 9). Nevertheless, their use was originally limited because single variable domains rarely retained the affinity of the parent antibody and were also poorly soluble and prone to aggregation.
The discovery that camelids (camels and llamas) and cartilaginous fish (wobbegong and nurse sharks) evolved into high-affinity, single, V-like domains (called VhH in camelids and V-NAR in sharks), mounted on an Fc-equivalent constant domain (8, 10-13), has resulted in translation of these camel, llama, and shark sdAbs into novel therapeutics. Camelid VhH and shark V-NAR domains display long surface loops, often larger than human antibodies and are able to penetrate cavities in target antigens (14). They are, in general, soluble and can be produced as stable I targeting reagents for sensitive diagnostic platforms and nano-sensors (14-17). However, for in-vivo administration, immunogenicity concerns require their humanization even though llama VhH domain have been claimed to be only minimally immunogenic (18). Thus, human single domains would be preferable for in-vivo applications provided problems of poor stability and solubility can be overcome.
Crystal and solution structures of several sdAbs adopt a β-sheet structure similar to a VH or VL immunoglobulin fold in a conventional antibody. sdAbs have similar affinity to antigens as whole antibodies, but are more heat-resistant and stable towards detergents and high urea concentrations. Those derived from camelid and fish antibodies are less lipophilic and better soluble in water, owing to their complementary determining regions (CDR3) that form an extended loop covering the lipophilic site that normally binds to a light chain (19, 20). Examples include the camelid-origin Nanobodies that consist of a single variable domain and two constant domains CH2 and CH3 (Ablynx). In contrast to common antibodies, two out of six sdAb survived a temperature of 90 C without losing their ability to bind antigens (21), a property that is attributed to a reversible unfolding behavior (7). Stability towards gastric acid and proteases depends on the amino-acid sequence. Some species have been shown to be active in the intestine after oral application, but their low absorption from the gut impedes the development of systemically active sAbs (22-26). The comparatively low molecular weight leads to a better tissue permeability, but also to short plasma half-lives and renal excretion (22).
Production of stable, high-affinity mAb fragments in high yield for preclinical and clinical trials can be a serious bottleneck in the product pipeline. Identification and design of mutations that minimize the hydrophobic interface and direct selection from phage libraries have contributed greatly towards overcoming solubility and stability problems (18-21). Bacteria are favored for expression of V-like domains, while human sdAbs can be expressed at high yields (> 50 mg L-1 in Escherichia coli and > 0.5 g L-1 in Pichia pastoris shaker flask cultures) (4). Nevertheless, several strategies are developed to improve recombinant expression such as the use of terminal polypeptides (e.g., c-Myc, histidine, and the “flag” epitope, DYKDDDK) to enhance affinity purification after expression into the periplasm of E. coli (4).
Immunogenicity concerns in relation to these strategies are still under investigation by drug regulatory agencies. Favorable properties, such as good expression, thermal stability, and solubility, are co-selected with binding activity using phage libraries, such as in the case of human VH domain antibodies (21), a method that enables also cost-effective production (see Table I).
Given their high affinity and specificity, the small size of sdAbs might make them particularly suitable for targeting antigens in obstructed locations, such as tumors where penetration into poorly vascularized tissue is crucial to the success of the drug (7). The delivery of toxins or radioisotopes to diseased tissues would be another ideal function for a sdAb, which could provide specific delivery of the toxin to the tumor while minimizing the length of time that the toxin could cause damage to healthy cells in the blood (7). The short half-life of sdAbs is well suited to applications in which rapid clearance is essential.
Long circulation half-life
For some applications such as treatment of brain disorders, rheumatoid arthritis, or cancers, the target antigens need to be available for binding in the bloodstream for a prolonged time requiring a prolonged serum half-life to increase time that the antibody has to act on its target and to reduce the frequency of administration (7). PEGylation, conjugation, or fusion to serum albumin, and fusion to either the Fc or complete antibody constant regions (7) have been employed; however, the latter method can lead to complement activation.
PEGylation increases the apparent hydrodynamic size of the antibody fragment above the glomerular clearance limit, thereby improving its circulating half-life, while also improving solubility, enhancing bioavailability by reducing losses at subcutaneous injection sites, and reducing immunogenicity and toxicity of therapeutic proteins (7). For example, conjugation of small human V domains (11-15 kDa) with a single PEG molecule extends the half-life from the normally rapid half-life of 20 minutes to 39 hours in mice (4). The site of modification needs to be carefully selected to ensure it does not interfere with the antigen-binding site. Conjugation/linkage of the sdAb fragment to a protein that has a naturally extended serum half-life protein like serum albumin is another method to prolong serum half-life of sdAbs. Site-specific conjugation of a Fab fragment to serum albumin by maleimide crosslinking or generation of a bispecific Fab with specificity for serum albumin in one arm increased half life in rats (27). Fusion of peptides with high affinity for serum albumin to an anti-tissue factor Fab lead to 37-fold and 26-fold extension in rabbit and mouse serum half-life, respectively (7). Albumin is also known to accumulate in tumors (28) and in arthritic joints, potentially enabling a further level of targeting in disease-specific situations (29). A range of human sdAbs isolated by phage display that bind to mouse, rat, and/or human serum albumin were produced that have half-lives that match the half-life of serum albumin (~19 days) (AlbudAbs) compared to a terminal half-life of non-serum albumin binding sdAbs of less than 45 minutes (29). The fusion of an Albu/Ab to interleukin-1 receptor antagonist (AlbudAb/IL-1ra) resulted in improved in-vivo efficacy due to its extended serum half-life (29). AlbuAbs could be used to generate a range of long half-life versions of many drugs (such as methotrexate, interferon alpha, peptide antagonists of TNFa) (13) to reduce frequency of administration and improve clinical profiles (Domantis-GSK, Phase I clinical trials, PN0621).
Applications in brain diseases
Due to prolonged aging, the prevalence of neurological disorders is growing, placing neurological disorders as a public health priority and an important cause of mortality constituting 12% of total deaths globally (30). The capillaries of the brain have evolved to constrain the movement of molecules and cells between the blood and brain, providing a natural defence against circulating toxic or infectious agents but also an effective barrier to drug delivery excluding 98% of therapeutic drugs. The relative impermeability of the blood brain barrier (BBB) results from tight junctions between capillary endothelial cells that are formed by cell-adhesion molecules, high expression of active efflux transport proteins, and large densities of mitochondria within endothelial cells (31). Thus, nearly all large molecules (molecular weight >1 kDa) such as recombinant proteins or gene-based medicines are unable to cross the BBB.
Conventional antibodies are unable to cross the BBB. Antibodies (and other peptides, proteins, and nucleic acids), however, have been shown to be able to be transported across by binding to receptors selectively expressed on brain capillary endothelial cells such as transferrin or the insulin-like growth factor (IGF-1) by receptor-mediated endocytosis (32). An alternative strategy for enhancing their transport relied on adsorptive-mediated endocytosis of cationized antibodies prepared by conversion of the surface carboxyl groups of the antibody to amino groups (33-35). However, even if an antibody is able to permeate the BBB, it will likely be the target of brain endothelial cell metabolism. Thus, the stability advantages of sdAbs (e.g., high expression levels, high affinity, high solubility, small size, and good intracellular stability) make them ideal for central nervous system therapeutic or imaging applications (see B).
Ablynx (Ghent, Belgium) is focused on the discovery and development of sdAbs, the Nanobodies, for a range of human diseases including inflammation, thrombosis, oncology, and neurology with seven sdAb products in the clinic (three in Phase II and four in Phase III) (36, 37). BI 1034020 Nanobody is being developed in collaboration with Boehringer Ingelheim for Alzheimer’s disease and, currently, is undergoing Phase I studies. Bi-paratopic humanized half-life extended anti-aβ VhH were shown to be able to bind to two different epitopes on the free aβ peptide (N-terminus and central) with an extremely high affinity (≤ 1 pM), reducing aβ levels in plasma in a preclinical model for Alzheimer’s disease (amyloid precursor protein [APP] transgenic mice) (38), which is associated with prevention of amyloid plaque formation in the brain and clearance of existing plaques (37). Ablynx is collaborating with Merck and Merck Serono to co-develop other sdAbs for neurological or neuro-oncological indications.
Transport across the BBB in human brain endothelial cells has been reported for llama sdAbs (39, 40). Using a llama sdAb phage display library (41), a new antigen-ligand system was identified for transvascular brain delivery (42). In-vitro and in-vivo studies have been reported focused on the development of two sdAbs, FC5 and FC44 (39, 43, 44), which selectively recognized human cerebrovascular endothelial cells and transmigrated across the BBB. The transport of FC5 and FC44 across the human brain endothelial cells is polarized, charge-independent, and temperature-dependent, suggesting a receptor-mediated process.
Abulrob et al. demonstrated that the transcytosis of FC5 is dependent on clathrin-coated vesicles and on the recognition of specific oligosaccharide antigenic epitopes on the surface of the human endothelial cells (39, 40). Following the internalization, FC5 was targeted to early endosomes, bypassed late endosomes/lysosomes, and remained intact after the transcytosis. FC5 failed to recognize brain-endothelial cells-derived lipids suggesting that it binds luminal α(2,3)-sialoglycoprotein receptor, which triggers clathrin-mediated endocytosis (39). Moreover, Abulrod et al. also described the modification of the FC5 sdAb with a horseradish peroxidase (HRP)-tagged IgG to improve its potential as a BBB carrier vector (40). Briefly, a free cysteine was engineered at the C-terminal of FC5, 17 amino-acid residues distant from the antigen-binding site. The cysFC5 maintained the transmigration ability across an in-vitro BBB model. The cysteine group was used further to attach the HRP-IgG (~190 kDa). It was shown that only HRP-IgG conjugated to the FC5 transmigrated across the in-vitro BBB model, suggesting that cysFC5 is able to transport larger molecules into the target tissue. The receptor has been identified, and is related to a novel isoform of the transmembrane domain protein 30A (TMEM 30A) (31). TMEM30A is also known as CDC50A, which is responsible for the cell surface expression of ATP8B1 (hypothesized to be a flippase for phospatidylserine). FC5 is now being developed to deliver therapeutic amounts of doxorubicin to the brain after pentamerization and association to PEGylated liposomes (31). Functionalization of nanoparticles with sdAb such as FC5 increases their valency, as multiple number of antibodies can be conjugated to the nanocarriers by spontaneous binding of the histidine tag of the antibody to the nitrilotriacetic acid (NTA) of the lipid. After intravenous administration (8.9 mg kg-1), brain levels of FC5-targeted, doxorubicin-loaded, PEGylated liposomes were increased (~0.6 µcg g-1 brain, double those achieved by doxorubicin-loaded, PEGylated liposomes). The pentameric form of FC5 (P5) displays better binding to brain endothelial cells and is more efficiently transcytosed across brain endothelial cells than the monomeric form and is co-localized with neurons in brain tissue (see Table II).
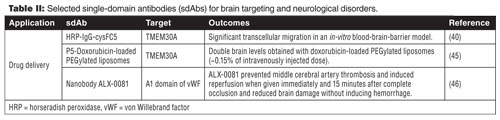
ALX-0081, a nanobody against the A1 domain of von Willebrand factor (vWF) that blocks vWF binding to main platelet receptor glycoprotein (GPIb), was compared with thrombolytic agent recombinant tissue plasminogen activator (rtPA) and tirofiban (glycoprotein IIb/IIIa inhibitor, antiplatelet agent) in a middle cerebral artery (MCA) guinea pig thrombosis model. ALX-0081 prevented MCA thrombosis and induced reperfusion when given immediately and 15 minutes after complete occlusion and reduced brain damage without inducing hemorrhage, whereas tirofiban prevented thrombosis but did not induce reperfusion and induced striking brain hemorrhage. rtPA also induced reperfusion when given 60 minutes after occlusion but provoked brain hemorrhage (see Table II). Skin bleeding time was not modified or was moderately prolonged by ALX-0081, whereas tirofiban and rtPA prolonged it. Thus, the inhibition of the GPIb-vWF axis by nanobodies in guinea pigs prevented cerebral artery thrombosis and induced early reperfusion without provoking intracerebral bleeding, hence, reducing brain infarct area; ALX-0081 administration, early after onset of an acute stroke, may induce revascularisation and reduce brain damage with less haemorrhagic risk than rtPA.
Conclusion
After a decade of intensive engineering followed by preclinical and clinical testing, antibody fragments joined mAbs as powerful therapeutic agents particularly for targeting cancer, inflammatory, autoimmune, and viral diseases. With recent advances in scaffold design, construction, and selection methodologies, there is now a rapid process for recombinant synthesis of specific, high-affinity antibody fragments against virtually any target. sdAbs are able to penetrate antigen clefts (e.g., enzyme active sites, cell surface receptors) targeting both antigens and immunosilent epitopes. sdAbs have shown impressive tumor-to-blood ratios and are increasingly applied towards the discovery of new cancer or inflammatory biomarkers and nanosensors while they are currently explored as highly refined biopharmaceutical drugs in challenging therapeutic areas such as neurology and oncology.
References
1. A. Beck et al., Nat Rev Immunol, 10 (5) 345-52 (2010).
2. J.M. Reichert, mAbs, 2 (1) 84-100 (2010).
3. J.M. Reichert et al., Nat Biotechnol, 23 (9) 1073-78 (2005).
4. P. Holliger and P.J. Hudson, Nat Biotechnol, 23 (9) 1126-36 (2005).
5. A.M. Wu and P.D. Senter, Nat Biotechnol, 23 (9) 1137-46 (2005).
6. E.S. Ward et al., Nature, 341 (6242) 544-46 (1989).
7. L.J. Holt et al., Trends Biotechnol, 21 (11) 484-90 (2003).
8. D. Saerens, G.H. Ghassabeh, and S. Muyldermans, Curr Opin Pharmacol, 8 (5) 600-608 (2008).
9. D. Gussow et al., Cold Spring Harb Symp Quant Biol, 1989. 54, Pt 1, 265-72 (1989).
10. E. De Genst et al., J Biol Chem, 280 (14) 14114-21 (2005).
11. E. De Genst et al., J Biol Chem, 279 (51) 53593-601 (2004).
12. H. Dooley and M.F. Flajnik, Eur J Immunol, 35 (3) 936-45 (2005).
13. V.A. Streltsov et al., Proc Natl Acad Sci USA, 101 (34) 12444-49 (2004).
14. B. Stijlemans et al., J Biol Chem, 279 (2) 1256-61 (2004).
15. S.D. Nuttall et al., Proteins, 55 (1) 187-97 (2004).
16. D. Saerens et al., J Biol Chem, 279 (50) 51965-72 (2004).
17. M. Pleschberger et al., Bioconjug Chem, 15 (3) 664-71 (2004).
18. V. Cortez-Retamozo et al., Cancer Res, 64 (8) 2853-57 (2004).
19. E. Dolk et al., Appl Environ Microbiol, 71 (1) 442-50 (2005).
20. R.L. Stanfield et al., Science, 305 (5691) 1770-73 (2004).
21. R.H.J. van der Linden et al., Biochim Biophys Acta, 1431 (1) 37-46 (1999).
22. M.M. Harmsen and H.J. De Haar, Appl Microbiol Biotechnol, 77 (1) 13-22 (2007).
23. M.M. Harmsen et al., Vet Microbiol, 120 (3-4) 193-206 (2007).
24. M.M. Harmsen et al., Vaccine, 23 (41) 4926-34 (2005).
25. M.M. Harmsen et al., Vet Microbiol, 111 (1-2) 89-98 (2005).
26. M.M. Harmsen et al., Appl Microbiol Biotechnol, 72 (3) 544-551 (2006).
27. B.J. Smith et al. Bioconjug Chem, 12 (5) 750-756 (2001).
28. R.K. Jain, Cancer Res, 48 (10) 2641-58 (1998).
29. L.J. Holt et al. Protein Eng Des Sel, 21 (5) 283-288 (2008).
30. M. Masserini, ISRN Biochemistry online, DOI: dx.doi.org/10.1155/2013/238428, April 11, 2013.
31. R. Gabathuler, Neurobiol Dis, 37 (1) 48-57 (2010).
32. R.J. Boado et al., Biotechnol Bioeng, 100 (2) 387-396 (2008).
33. A.W. Vorbrodt, J Neurocytol, 18, 359-368 (1989).
34. W.M. Pardridge et al., J Pharm Sci, 84 (8) 943-8 (1995).
35. D. Triguero et al., Proc Natl Acad of Sci, 86 (12) 4761-4765 (1989).
36. K. Deffar et al., Afr J Biotechnol, 8 (12) 2645-2652 (2009).
37. Ablynx, Nanobodies, www.ablynx.com, accessed Jul 8, 2014.
38. J.E. Park et al., “Biparatopic Abeta Binding Polypeptides,” US patent WO2011/107507, filed Mar. 2011.
39. A. Abulrob et al., J Neurochem, 95 (4) 1201-14 (2005).
40. A. Abulrob et al., International Congress Series, 1277 (0) 212-223 (2005).
41. J. Tanha et al., J Immunol Methods, 263 (1-2) 97-109 (2002).
42. J. Tanha, A. Muruganandam, and D. Stanimirovic, Methods Mol Med, 89, 435-49 (2003).
43. A.S. Haqqani et al., Mol Pharm, 10 (5) 1542-56 (2013).
44. A. Muruganandam et al., FASEB J, 16 (2) 240-242 (2001).
45. A. Abulrob and J. Zhang, “Targeted delivery of compounds using multimerization technology,” Canada patent WO 2007/036022, filed Sept. 2006.
46. S. Momi et al., Blood, 121 (25) 5088-97 (2013).
About the Authors
 *Aikaterini Lalatsa, PhD, is a lecturer in pharmaceutics and drug delivery, email: katerina.lalatsa@port.ac.uk, tel.: +44 (0) 23 9284 3929; the School of Pharmacy and Biomedical Sciences, University of Portsmouth,
*Aikaterini Lalatsa, PhD, is a lecturer in pharmaceutics and drug delivery, email: katerina.lalatsa@port.ac.uk, tel.: +44 (0) 23 9284 3929; the School of Pharmacy and Biomedical Sciences, University of Portsmouth,
St Michael’s Building, White Swan Road, Portsmouth PO1 2DT, United Kingdom.
*To whom all correspondence should be addressed.
 Diana M. Leite is a PhD student at the School of Pharmacy and Biomedical Sciences, University of Portsmouth, St Michael’s Building, White Swan Road, Portsmouth PO1 2DT, United Kingdom.
Diana M. Leite is a PhD student at the School of Pharmacy and Biomedical Sciences, University of Portsmouth, St Michael’s Building, White Swan Road, Portsmouth PO1 2DT, United Kingdom.