Stage 2: Criticality and process qualification
The FDA Process Validation Guidance requires qualification in that “utilities and equipment are suitable for their intended use” as part of Stage 2 (process qualification) (1). Suitability, or fit for purpose, is assessed either through risk-based commissioning and qualification (e.g., ASTM E2500 [6]), or through the traditional installation, operation, and performance qualification approach.
The acceptance criteria for qualification of equipment and utilities must be consistent with the Stage 1 control strategy. Operational qualification studies must show that the utilities/equipment is capable of controlling each relevant CPP throughout its NOR. The risk level of the CPP is used to determine the amount of testing so that high-risk CPPs require more replication and rigorous data analysis and low-risk CPPs require a simple verification. Equipment performance qualification can be coordinated with full-scale process characterization studies to test the manufacturing equipment under representative product conditions with the CPPs up to the limits of their NORs. These full-scale studies also provide the opportunity to confirm the suitability of planned sampling plans and acceptance criteria for process performance qualification (PPQ).
The continuum of criticality also influences the study design for PPQ including number of batches, sampling plans, and study acceptance criteria. Under the new FDA Process Validation Guidance, the purpose of the PPQ is to demonstrate that the process design and control strategy are capable of meeting CQA acceptance criteria for not just a fixed number of PPQ lots, but for future commercial lots (1). For this reason, while releasing the defined number of PPQ lot under protocol is certainly a necessary criteria, it is not sufficient in and of itself to provide assurance that the process is under control and will continue to produce releasable future lots. Trying to qualify an out-of-control process creates a possible scenario of having the individual PPQ lots pass their acceptance criteria (i.e., they each meet specifications) but the process performance qualification criteria do not pass.
Since the PPQ is a means of confirming process reproducibility under typical production conditions, CPPs and KOP are expected to be set at their normal set points and remain within their NOR. Studies executed at the limits of NOR ranges are done during Stage 1 (process design) and must be completed before Stage 2 (process qualification).
The number of PPQ lots and the study acceptance criteria are linked by both statistical and risk-based analysis of process characterization (Stage 1) data and existing process knowledge. Both the Parenteral Drug Association (PDA) Technical Report 60 (7) and the International Society for Pharmaceutical Engineering (ISPE) Product Quality Lifecycle Initiative (PQLI) guide series (8-10) provide several example methods and are excellent resources on the topic. Some possible choices for determining the number of lots are:
• Structural: determined by process complexity, dosage form strengths, and number of equipment trains; this includes bracketing and matrix strategies and may involve separating groups of unit operations into separate PPQ protocols.
• Risk-based: uses a comprehensive analysis to assess how much process risk remains after applying existing process knowledge and process design data.
• Statistical: based on calculations targeting capability, tolerance intervals, or overall reliability of meeting CQA acceptance criteria.
Companies may combine these strategies with each other or with business requirements to produce sufficient batches for launch quantities. These strategies for selecting the number of PPQ lots and the specific steps used are described in or referenced by the process validation plan.
Where practical and meaningful, a statistical method of determining the number of batches is recommended, although there is no standard industry approach. Statistical methods inherently incorporate the risk component reflecting the level of understanding derived from Stage 1 of the process validation lifecycle. Misapplication of these risks may lead to unjustified confidence in PPQ batch results. The statement “all five PPQ lots pass CQA acceptance criteria” has no statistical meaning in determining the amount of risk the process control strategy has in producing future successful lots. A statistical criteria such as “results from the five passing PPQ lots will show that 90% of future lots are expected to meet CQA acceptance criteria with a 95% confidence” describe a well-controlled process that not only will produce five successful lots, but also is highly likely to produce successful lots in the future.
It is generally difficult to prove tight statistical criteria (90–95% confidence and 95–99% conformance or coverage) with PPQ lots alone. One strategy is to apply wider criteria (50% confidence) to a smaller number of PPQ lots with monitoring of a larger number of CPV (Stage 3) lots to meet the target statistical criteria (95% confidence). These CPV lots use the same enhanced sampling and monitoring as the PPQ lots, but are released under normal batch acceptance criteria. A statistical approach to determining the number of PPQ batches is often used in combination with risk-based or structural strategies.
Per the FDA Process Validation Guidance, sampling plans for PPQ must be “more extensive than is typical during routine production” and “be adequate to provide sufficient statistical confidence of quality both within a batch and between batches. The confidence level selected can be based on risk analysis as it relates to the particular attribute under examination.” Since the continuum of criticality has been applied to CQAs and in-process controls, the risk level may be used to determine acceptance criteria and sampling requirements.
The following example uses statistical tolerance intervals (11) where coverage is the proportion of the expected “future” data to be contained within the acceptance limits:
• High-risk CQA: 99% coverage at 95% confidence
• Medium-risk CQA: 95% coverage at 95% confidence
• Low-risk CQA: 90% coverage at 95% confidence.
The actual number of samples required varies based on the data type (discrete or continuous) and the expected distribution (normal or unknown). The samples are assessed by individual lot, or if no significant lot-to-lot variation is seen, by pooling the sample data across lots.
Stage 3: Criticality and continued process verification
At the conclusion of a successful PPQ, process validation activities move into an ongoing monitoring and review phase called continued process verification (CPV). It should now be clear that each of the previous two process validation stages have limitations. Stage 1 (process design) depends upon risk analysis, prior knowledge, and scientific principles, because not all possible parameters and interactions can be evaluated through experimentation. Many design of experiments (DOEs) are performed on smaller scale with limited material variation to conserve resources. Stage 2 (process qualification) has a limited number of commercial-scale runs to provide confidence in the control strategy developed in Stage 1 and cannot fully explore all raw material variability. PPQ studies can only provide a limited amount of statistical confidence that the CQAs will continue to meet their acceptance criteria in the future.
Continued process verification is the recognition that process validation is a lifecycle that does not end with PPQ and the start of commercial production. In exchange for CPV, FDA will allow changes to a process without revalidation when process drift is observed for CPPs that have been identified so long as they do not violate the defined PAR and are based upon Stage 1 and Stage 2 process understanding.
Despite best efforts, the design and control spaces are models limited by the data and circumstances from which they are developed. Process parameter and material attributes determined to have little or no impact may undergo sudden or subtle shifts, which may drive CQAs beyond their acceptance limits. CPV is an ongoing program for monitoring and statistical analysis of these critical inputs and their expected relationship with CQAs as defined by the design space.
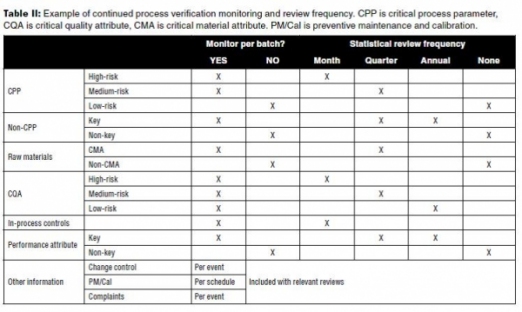
Table II is an example of the monitoring and review frequency for a process based on the established continuum of criticality for parameters and attributes from Stages 1 and 2. In this example, low-risk parameters (such as non-critical, non-key) are not monitored due to their low impact on quality attributes. They may, however, be initially monitored for several of the first commercial lots following PPQ to confirm their low impact. The review frequency shown in the table is built on an assumption of a frequently manufactured product such that each review includes at least several lots of new data.