The authors discuss complications of implementing continued process verification and provide recommended approaches.
Oct 1, 2014
By: Jeff Fleming, Robin Payne
BioPharm International
Volume 27, Issue 10, pp. 48-52
 Continued process verification (CPV) is the activity that provides ongoing verification of the performance of a manufacturing process. Guidance issued by FDA in 2011 (1) emphasized the importance of manufacturers undertaking CPV as an integral part of the process validation lifecycle. CPV will provide the manufacturer with assurance that a validated process remains in a validated state during the routine manufacturing phase of the product lifecycle.
Continued process verification (CPV) is the activity that provides ongoing verification of the performance of a manufacturing process. Guidance issued by FDA in 2011 (1) emphasized the importance of manufacturers undertaking CPV as an integral part of the process validation lifecycle. CPV will provide the manufacturer with assurance that a validated process remains in a validated state during the routine manufacturing phase of the product lifecycle.
A group of more than 20 companies collaborated on this topic, using the facilitation services of the BioPhorum Operations Group (BPOG), to generate a detailed example of how CPV for a biopharma manufacturing process might be implemented and explaining the thought process behind the development of a CPV plan (2). Its recommendations are based on a typical cell culture production process for making a fictitious monoclonal antibody (mAb) product (3). Although this model plan does not provide examples of all aspects that may be included for other types of processes, the concepts and principles upon which the content was derived should help with CPV implementation for any real product. Preparation of the CPV plan has provided a basis upon which to build and share knowledge and initiate further clarifying discussion across the industry.
What is Continued Process Verification?
CPV is fundamentally a formal means by which the performance of a commercial manufacturing process is monitored to ensure consistently acceptable product quality. In its entirety, CPV includes: preparation of a written plan for monitoring a biopharmaceutical manufacturing process, regular analysis of results, documentation of the data collected, analysis of data, and actions taken based on the results of monitoring the process. Some elements of CPV overlap with existing GMP systems such as the generation of data for batch release (BR) decisions, annual product review (APR), and change control.
In general, the nature and extent of CPV should be aligned with the outcomes of process qualification by focusing attention on aspects of processing that are most important to determining the quality attributes of the product. Limiting the focus of CPV to product quality attributes is important to assure that the burden of data collection and analysis does not grow too large. Simply put, if a measurement cannot be associated with one or more product quality attributes, it is not essential for CPV. A manufacturer may also wish to include measures of process efficiency, but beyond these two categories, adding additional measures to those taken during processing will not add significant value to the performance monitoring task.
Table I is a sample showing the review of variables for inclusion in the model CPV plan for the A-mAb (the conceptual mAb described in references 2 and 3) production-scale bioreaction step. Only some of the variables considered for this step in the model CPV plan are shown in Table I. As indicated, some variables were not selected for CPV or were made optional because it was felt the variables selected for CPV are sufficient to assess process performance and consistency, while other variables may not be appropriate for monitoring process performance and/or consistency. In practice, the selection of variables for CPV should be determined on a case-by-case basis by those with expertise in the process and with statistical evaluations. For the complete set of tables for this process, see Reference 2.
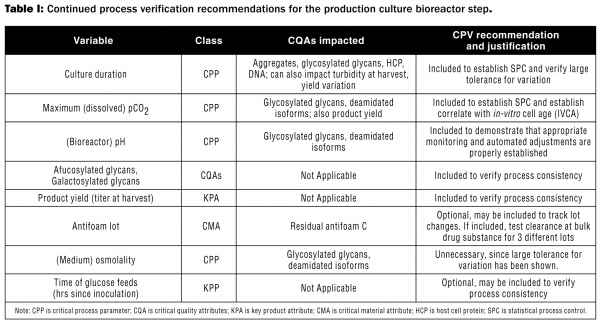
While a CPV plan will include analysis of data related to BR, CPV operates independently from the BR process and should not have any impact on batches that have been previously released. Data and analyses provided by CPV should be used to meet APR requirements, eliminating redundancy between these GMP systems.
CPV and Quality by Design
Quality by design (QbD) means scientifically building quality into a product and the process by which it is developed and manufactured. QbD is based on the systematic identification of the risks involved in manufacturing the product and mitigation of those risks. QbD recognizes that scale-up and industrial-scale commercial manufacturing experience provide knowledge about the process and the suitability of raw materials used. FDA’s January 2011 process validation guidance emphasizes the need for companies to organize and use acquired knowledge and continually improve throughout the process lifecycle by making adaptations to correct root causes of observed manufacturing problems (1).
By providing the mechanism for ongoing acquisition of process knowledge, CPV provides an optimal mechanism for achieving the goal of measuring process performance and, therefore, fits well within the QbD framework. CPV provides the essential link in the product lifecycle chain by augmenting knowledge gained during the product development phase with that acquired during commercial product manufacturing phase. CPV serves to provide ongoing verification of the process design and aids in enhancement of process understanding providing the vital link shown at the left side of the inner circle in Figure 1.
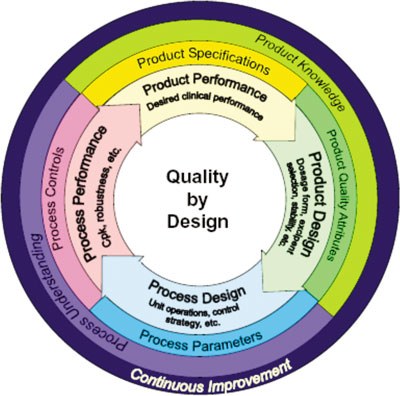 Figure 1. Elements of the product lifecycle (4). The pink zone indicates where continued process verification plays its part.
Figure 1. Elements of the product lifecycle (4). The pink zone indicates where continued process verification plays its part.
While in some cases data from QbD may be available and advantageous, preparing and executing a CPV plan does not necessitate a manufacturing process is developed using a QbD approach. Indeed, there may be times when limited development history (e.g., as may be the case for some legacy and in-licensed products) makes it necessary to create a CPV plan without a QbD background. The CPV plan can be tailored to focus mostly on performance of the process at the time of qualification/validation, the manufacturing history to the time a CPV plan is created, and expectations for the process from that point forward.
Initial and Long-term Control Limits
Control limits for CPV should be based on process data. Depending on the extent of experience with the process, different limits for both an initial monitoring period and long-term manufacture may be advisable for CPV. For a newer process, because it is unlikely that there will be a statistically robust set of data at the scale of commercial manufacture, it is recommended that an initial phase of CPV be established. Initial limits will typically be based on prior process experience (e.g., from the process validation campaign, even if this is limited) and data from the development phase. Data gathered during the initial monitoring period will then help to establish limits that represent the performance of the process at the commercial scale.
In general, statistical control limits should be set at the centerline plus and minus three sigma, which approximates to three standard deviations. Sigma may be estimated from short-term or long-term variation using the established formulae.
The long-term variation version of sigma is preferred for two reasons. First, it is more likely to include all the sources of variation for the process. This provides more realistic control limits that will be better able to distinguish between expected and unusual instances of variation in results. And secondly, during initial production, when relatively few results are available for calculating sigma, the long-term estimate stabilizes more quickly than the short-term estimate. For independent, normally distributed results, the long-term and short-term formulas for sigma will provide similar values.
After the initial phase of CPV, control limits should be established against which the performance of the process can be assessed for the longer term. These control limits are expected to be more statistically reliable and should not be changed without recorded justification. As a rule of thumb, 30 batches worth of data might be expected to reflect all sources of variation in the process, but this must not be regarded as a hard and fast rule. When sources of variation occur over extended timeframes, more than 30 results may be needed to reflect all sources of variation. And, there are some sources, like raw material changes, that occur quite infrequently, making it important to monitor data on an appropriate frequency at different levels in the organization. Typically, operators will monitor data during manufacture and review the data at the end of a campaign for infrequently manufactured products or weekly/monthly for frequently manufactured products, with a quarterly report that contributes to the annual product report (APR).
A CPV plan should be regarded as a living document that is updated accordingly, as process changes are made. For instance, when a process change or improvement shifts the mean or changes the variability, control limits should be re-set based on an appropriate number of results following the change.
The Rewards of Process Performance Monitoring
CPV provides the opportunity to identify and control sources of variation and, therefore, improve process robustness and increase assurance of reliable supply to the market. Adopting or building on an existing system of monitoring manufacturing process performance is likely to involve collection of considerably more data over the lifetime of the product than may have occurred previously. Analysis of data and reporting for CPV may also involve examination of existing process measurements and improved methods for data tracking and analysis beyond what is typically done for process validation. The reward for this additional data collection and analysis is that CPV will provide information from which to improve process understanding, risk assessment/mitigation, and the control strategy (CS), and will generate an established foundation for justifying process improvements throughout the lifecycle of the product. Of course, having and following a CPV plan should also put the manufacturer in more favorable standing with GMP regulatory inspectors.
One of the main struggles to resolve when implementing CPV will be to determine the types and quantity of data to be captured and analyzed. CPV can err on the side of providing too little data to the manufacturer, or can lead to the over-collection of unimportant data that masks the valuable stuff and adds to the costs incurred in interpretation. Decisions on data capture and analysis will always have to be determined on a case-by-case basis. But with careful forethought, CPV can fulfill both regulatory needs and provide value to the manufacturer.
The implementation and ongoing execution of a CPV Plan is likely to require additional effort beyond what is typically needed to prepare the APR, because CPV reports are issued more frequently than the APR. The data presented in a CPV report will have a degree of overlap with that for the APR, but a direct relationship in the data contained is unlikely, as these reports meet somewhat different purposes. Additional cost may be incurred due to the need for quality risk assessments to define the scope and frequency of data collection and the time and effort needed to collect and analyze more data. The frequency of collection and analysis will depend on several factors, including: whether production is campaigned or continuous; variability relative to process control limits an statistical capability (e.g., high capability for a parameter can justify reduced monitoring of the parameter); whether risks to product quality (and thus product disposition) and process consistency are sufficiently mitigated; and the intended use of the reported data (e.g., use in continuous improvement may mean collecting and analyzing certain data for each batch produced).
Future Efforts and Improvement
The BPOG CPV example plan prepared by the collaborative team does address some of the complications of implementing CPV and provides recommended approaches, but questions related to information technology (IT) systems are not dealt with in any detail. The case for IT systems (including their design, introduction, and possible efficiency advantages) is the subject of ongoing collaborative efforts facilitated by BPOG; results of that work may be published in the future.
References
1. FDA, Guidance for Industry, Process Validation: General Principles and Practices (CDER, 2011).
2. BPOG, Continued Process Verification: An Industry Position Paper with Model Plan, accessed Sept. 16, 2014.
3. CMC Biotech Working Group,A-Mab: a Case Study in Bioprocess Development(Oct. 30, 2009), accessed Sept. 16, 2014.
4. From presentation at 10th APIC/CEFIC meeting, “FDA’s Quality Initiatives—An Update” by Moheb M. Nasr
About the Authors
Jeff Fleming is an independent writer with over 30 years of experience in pharmaceutical manufacturing.
Robin Payne is facilitator at the BioPhorum Operations Group, robin@biophorum.com .